











Medizintechnik | Sicherheit
ERROR: FEHLERHAFTE IMPLANTATE
Seite 1/1 2 Minuten
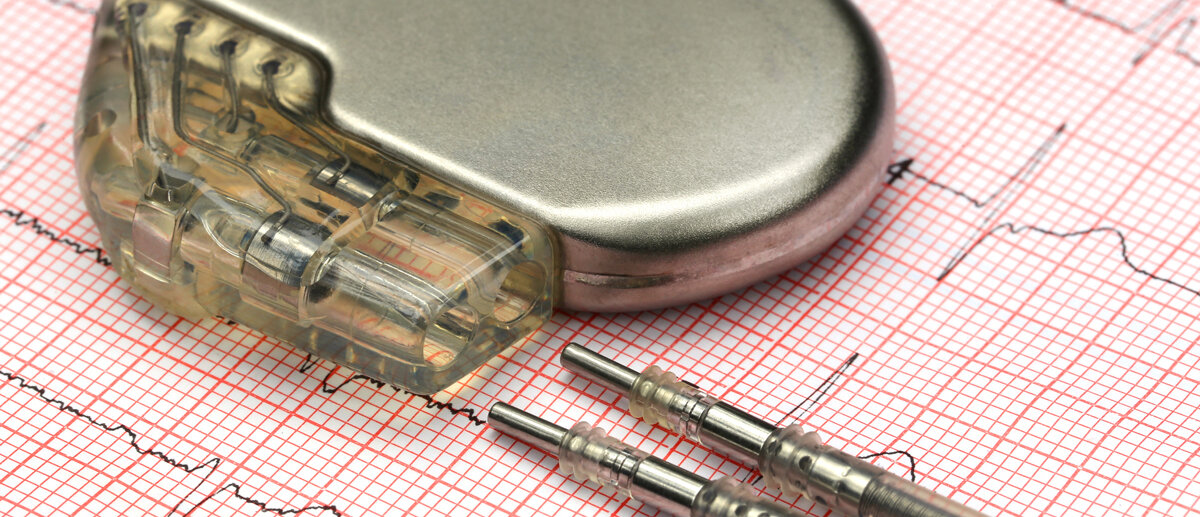
Nach Angaben der Recherchegruppe seien in Deutschland im vergangenen Jahr über 14 034 Fälle gemeldet worden, bei denen es zu Verletzungen, Todesfällen oder anderen Problemen gekommen sei, die im Zusammenhang mit Medizinprodukten stehen könnten – wobei die Verdachtsfälle immer weiter zunähmen. Dabei dreht sich die Kritik hauptsächlich um die Medizinprodukte der Risikoklasse III, also um solche, die direkt an Gehirn oder Herz angewendet oder in den Körper gesetzt werden wie etwa Herzschrittmacher, Herz-Lungen-Maschinen, Prothesen oder Brustimplantate. Demnach seien in deutschen Krankenhäusern 3170 Implantate wieder herausoperiert worden, weil das Gewebe rund um die Silikonkissen schmerzhaft vernarbt gewesen sei. Gemeldet wurden davon wohl nur 141 Fälle.
Das BfArM verkündete wiederum indessen ein zunehmendes Meldeverhalten von Ärzten und Kliniken, diese werden auch regelmäßig vom BfArM auf die Bedeutung der Meldungen hingewiesen. Darin sieht das Institut auch einen Grund für die Zunahme der registrierten Fälle, zudem steige die Zahl der auf dem Markt befindlichen Medizinprodukte. Die aktuelle Zahl der gemeldeten Fälle beträgt laut der Homepage des BfArM 12 000 (Stand 2016). Wobei das Institut nach eigener Darstellung bei seinen Überprüfungen festgestellt hat, dass in rund 40 Prozent der Fälle nicht das Medizinprodukt Schuld an einer Verschlechterung des Gesundheitszustands des Anwenders gewesen sei.
In „Implant Files“ wird auch der Ablauf der Zulassung kritisiert. Das Problem sei, dass die Medizinprodukte nicht von staatlichen Stellen kontrolliert und zertifiziert werden müssten, sondern private Institute im Auftrag des Herstellers prüfen und ein CE-Siegel vergeben. Dadurch kann es in der gesamten EU verkauft werden. Auf seiner Homepage erklärt das BfArM das Zulassungsverfahren von Medizinprodukten folgendermaßen: Je nachdem welche Risikoklasse vorliegt wird auch das Konformitätsbewertungsverfahren durchgeführt. So können sich Hersteller bei Risikoklasse I in Eigenverantwortung bewerten, wohingegen bei höheren Risikoklassen unabhängige Prüf- und Zertifizierungsstellen hinzugezogen werden, die durch den Staat benannt werden und in Deutschland von der Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten (ZLG) benannt und überwacht werden. Der Hersteller wählt dann eine solche sogenannte Benannte Stelle frei aus.
Auch der Bundesverband Medizintechnologie (BVMed) meldete sich heute zu Wort und wies daraufhin, dass die MedTech-Branche durch die neue EU-Medizinprodukte-Verordnung stark reguliert sei und hohe Anforderungen an die Benannten Stellen gestellt seien. Dabei hat erst vergangenen Monat das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) bemängelt, dass diese Neuregelungen zu kurz greifen würden, vor allem wenn es um neue Behandlungsmethoden gehe, die auf der Anwendung von Hochrisikoprodukten beruhten.
Farina Haase,
Apothekerin, Volontärin
Quelle: dpa
Ärzteblatt
www.bfarm.de